Overview
What is GDEE?
Gene Discovery and Enzyme Engineering (GDEE) is a comprehensive Python package that provides the functionality necessary to run the Gene Discovery and Enzyme Engineering Platform developed under the project ShikiFactory 100 (European Union’s Horizon 2020 research and innovation programme under grant agreement number 814408).
GDEE integrates multiple computational approaches for protein engineering, enabling researchers to design, model, and evaluate protein variants through automated workflows.
How do I install GDEE?
GDEE requires several external software dependencies and Python packages. Installation involves setting up both the computational tools and the Python environment.
Prerequisites
Before installing GDEE, you need to install the following external software:
MODELLER: Required for homology modeling (academic license needed)
AutoDock Vina: For molecular docking calculations
Smina: Alternative docking tool with Vinardo scoring
MGLTools: For structure preparation and PDBQT file generation
VoroMQA: Optional, for model quality assessment using Voronoi analysis
Python Package Installation
GDEE can be installed directly from the source:
cd gdee/package
pip install .
Dependencies
GDEE automatically installs the following Python dependencies:
numpy (≥1.14) - Numerical computations
mdanalysis (≥0.20) - Molecular structure analysis
mpi4py (≥3.0) - Parallel computing support
path.py (≥13.0) - Enhanced path operations
biopython (≥1.75) - Biological sequence handling
oddt (≥0.7) - Cheminformatics toolkit
openbabel-wheel (≥3.1.1.1) - Molecular file format conversions
six (≥1.17.0) - Python 2 and 3 compatibility
scipy (==1.9.0) - Scientific computing
For detailed installation instructions, see the Installation and Setup guide.
How can I use GDEE?
GDEE provides a high-level Python interface through the ProteinEngineering class. Here’s a basic workflow example:
Basic Usage Example
from gdee import ProteinEngineering
# Initialize the protein engineering platform
engineer = ProteinEngineering("my_protein", "results.db")
# Set the template PDB structure
engineer.pdb = "template.pdb"
# Configure variant generation (mutation-based approach)
engineer.variant["name"] = "mutation"
engineer.variant["matrix"] = "blosum62"
engineer.variant["selection"] = "A:100 A:150 A:200" # Residues to mutate
engineer.variant["max_iterations"] = 100
engineer.variant["conservative"] = True
# Configure modeling parameters
engineer.model["name"] = "modeller"
engineer.model["num_models"] = 5
engineer.model["optimize_level"] = 1
# Add ligand for docking
ligand = engineer.add_ligand("substrate", "ligand.pdbqt")
# Add distance measurement between cofactor and ligand using MDAnalysis selection syntax
ligand.add_measurement("cofactor-ligand", "distance",
"chainId A and resid 195 and name N4", "name C1")
# Configure docking parameters
engineer.evaluator["name"] = "vina"
engineer.evaluator["box_center"] = [10.0, 15.0, 20.0]
engineer.evaluator["box_size"] = [20.0, 20.0, 20.0]
engineer.evaluator["exhaustiveness"] = 100
# Run the engineering campaign
engineer.run()
Key Configuration Options
Variant strategies: Choose between “mutation”, “msa”, or “exhaustive” approaches
Selection syntax: Specify residues using MDAnalysis selection strings
Quality thresholds: Set DOPE and VoroMQA cutoffs for model filtering
Parallel execution: Configure MPI settings for distributed computing
Output management: Control file organization and compression
Re-Scoring: Optionally re-score docking poses with trained metamodel for improved ranking
For comprehensive examples and advanced usage, see the Usage Instructions guide.
How does GDEE work?
GDEE implements a modular pipeline architecture that processes protein variants through multiple computational stages. The workflow is designed to be both automated and highly configurable.
Pipeline Architecture
The GDEE pipeline consists of the following sequential steps:
Variant Generation → 2. Structure Modeling → 3. Quality Assessment → 4. Molecular Docking → 5. Distance Measurements → 6. Data Storage
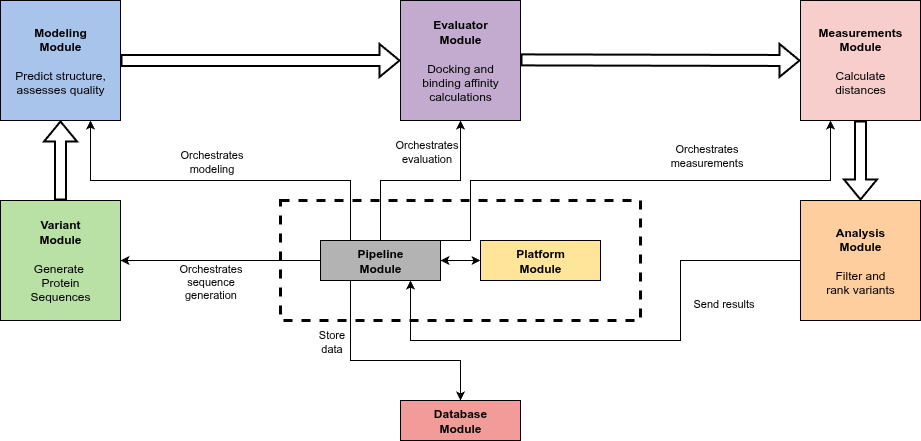
High-level diagram illustrating the architecture and data flow of the platform. The Core functions as the central orchestrator coordinating and scheduling task execution across specialized modules in a linear pipeline workflow – Variant, Modeling, Evaluator, Measurement, and Analysis – each dedicated to specific tasks on protein variants. The Variant module generates amino acid sequence variants using different strategies; the Modeling module predicts 3D structures and assesses their quality; the Evaluator module performs docking and binding affinity calculations; the Measurement module calculates distances between selected protein and ligand atoms to assist in filtering; and the Analysis module applies filters and ranks variants accordingly. Data flows between processing modules are represented by bold arrows, while narrow arrows indicate the Core orchestration and management of operations. All intermediate and final results are centralized in the Database module, providing efficient data storage and retrieval for downstream analysis.
Detailed Workflow
- Step 1: Variant Generation
GDEE supports multiple strategies for generating protein variants:
MSA-based variants: Generate variants based on FASTA file (Can result from a BLAST search - Gene Discovery)
Matrix-based mutations: Use substitution matrices (BLOSUM62 or custom) for guided mutations
Exhaustive combinatorial mutations: Systematically explore all possible combinations
Conservative vs. non-conservative mutations: Control mutation bias based on amino acid properties
- Step 2: Structure Modeling
Uses MODELLER to create 3D structural models for each sequence variant
Generates multiple models per variant to account for conformational uncertainty
Performs local optimization focused on mutated regions to minimize computational cost
- Step 3: Quality Assessment
Evaluates each model using Normalized DOPE scores from MODELLER
Optionally applies VoroMQA for additional quality validation
Filters out low-quality models based on user-defined thresholds
- Step 4: Molecular Docking
Prepares protein models in PDBQT format using MGLTools
Performs molecular docking using AutoDock Vina or Vinardo scoring
Generates multiple binding poses per protein-ligand complex
- Step 5: Distance Measurements
Calculates user-defined distance measurements between protein and ligand atoms
Stores distance measurements for each pose
- Step 6: Data Storage
Saves all results to a SQLite database with hierarchical organization
Archives output files (PDB models, docking poses) in compressed format
Maintains data integrity through transactional database operations
Supported File Formats
Input: PDB files for template structures (protein), PDBQT files for ligands
Output: PDB files for models, PDB files for docking poses
Database: SQLite format for persistent storage